ShiLiQiYe
Bioincloud
{示例企业}生科云
关
键词1
标语1.........(这是一段可以配置的话)
arrow_left
arrow_right
menu
生科云,AI4S探路者
首页
SVG编辑器
微信登录
账号登录
person
lock_open
云流程
云流程基本介绍
linear_scale
我的流程
查看和使用云流程
云工具
所有工具的索引
我的任务
云工具的任务状态
关于我们
{示例企业}介绍
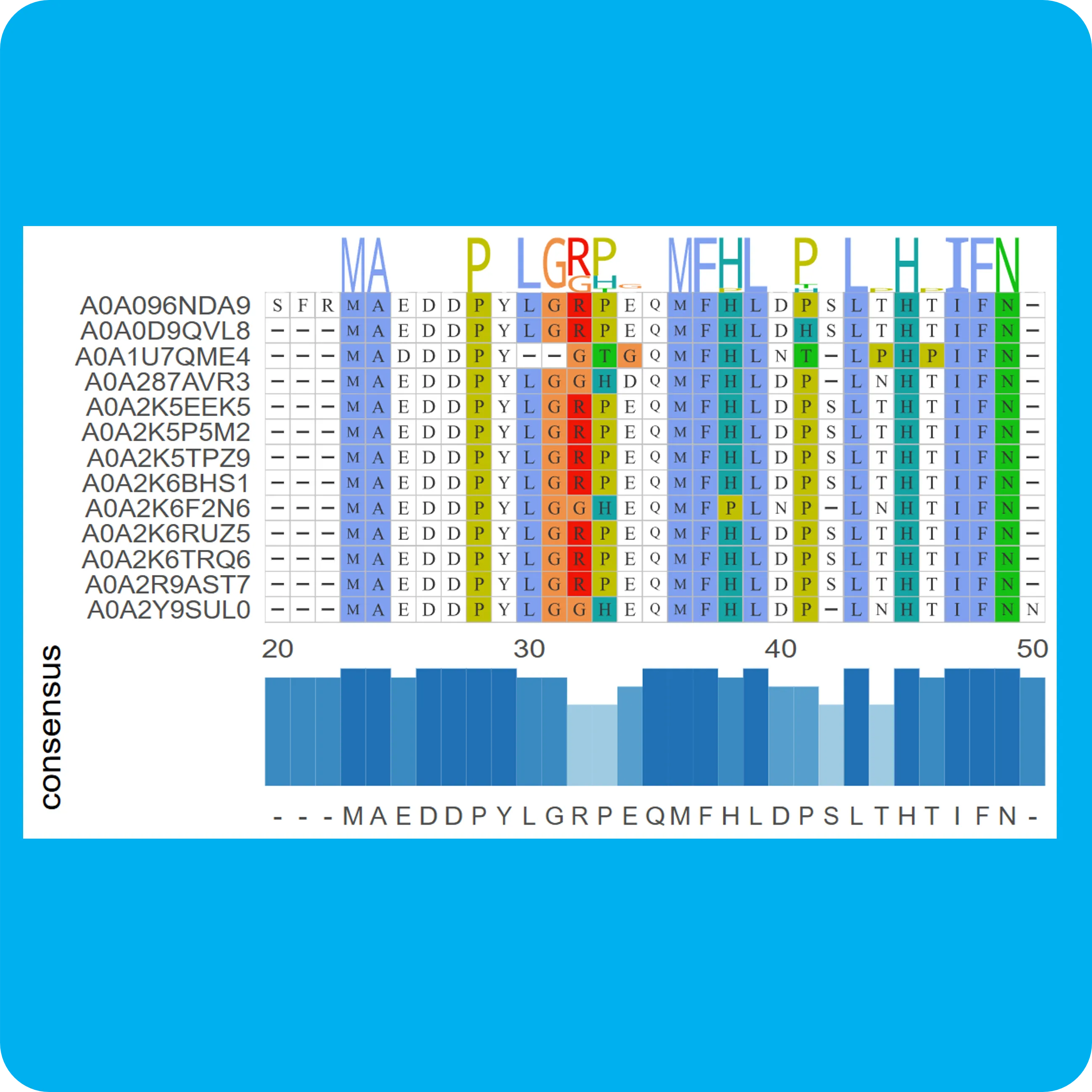
MSA多序列比对
使用FAMSA对fasta格式的多序列数据进行比对,使用R语言ggmsa包进行结果可视化,其结果用于分析多条蛋白质序列(或DNA,RNA)之间的同源性,比较序列差异,推断序列进化历史和进化方式(突变/插入缺失/倒置等)。
tips_and_updates
想了解更多?您还可以 查阅相关流程示例
结题报告
,或问问
{示例企业}AI
多序列数据:
help
第一行应为>开头的序列标识,唯一不可重复;第二行开始应为序列本身,可换行;接下来的序列也是如此;请输入.fasta格式的多序列数据,序列名称不可重复,每次输入的序列量应不大于20条
选择输入文件
folder_open
示例
cloud_download
开始位置:
help
设置序列开始位置
结束位置:
help
设置序列结束位置,结束位置需要大于序列开始位置
序列颜色:
help
ggmsa包提供了多种颜色选择,根据需要选择颜色类型,如果是核苷酸数据请选择末尾带有_NT的参数
Clustal
字体:
help
ggmsa包提供了多种字体选择,根据需要选择字体类型
TimesNewRoman
字体大小:
help
图像中表示氨基酸或者核苷酸的字母宽度
显示序列名:
help
选择是否显示序列名称,即序列文件中 > 符号后的内容
是
否
添加seqlogo:
help
选择是否添加seqlogo
是
否
添加msabar:
help
选择是否添加序列保守性柱状图
是
否
图像高度:
图像宽度:
任务备注:
help
0-20个字母,数字,下划线或汉字的组合
mail
任务完成时发邮件提醒我
提交任务
help
使用说明
加载中...